

缓生日本根瘤菌菌种间的共生固氮作用不同。接种USDA123的大豆产量低于已知固氮效率高的菌株,如USDA110。在美国中西部大豆主产区,USDA123在大田大豆中结瘤率高,具有一定的竞争力,但固氮效率较低。以1321份甘氨酸max和69份甘氨酸大豆为材料,分别接种USDA110和USDA123,建立了高通量的根瘤数鉴定体系。
鉴定出73份USDA110和USDA123根瘤数差异显著的G. max材料。对73份材料中的35份进行双接种后,发现PI189939、PI317335、PI324187B、PI548461、PI562373和PI628961均被USDA110和双株结核占据,而不被USDA123结核单独占据。PI567624仅被USDA110结核占据,PI507429限制所有菌株。结果表明,接种USDA110菌株和USDA123菌株分别有35个和35个位点与根瘤数相关。接种USDA110和USDA123分别鉴定出23个位点和34个位点。只有4个基因座在两个处理中是共同的,每个基因座只能解释0.8 - 1.5%的表型变异。
建立了表征根瘤菌数量和占用率的高通量表型系统,并鉴定出限制根瘤菌USDA123而偏爱USDA110的大豆种质。次要效应较多,控制结瘤数的共同位点较少,表明性状遗传复杂性和株系依赖性结瘤限制。本研究的信息将有助于开发限制USDA123的品种,从而提高固氮效率和生产力。
大豆[甘氨酸max (L.)]稳定。]是世界上最重要的经济作物之一,因其丰富的蛋白质和油脂含量,分别约为42%和20%[1,2]。大豆蛋白质含量对氮的需求量很大,主要通过生物共生固氮和土壤残氮库两种方式提供氮,其中缓生根瘤菌对大气氮具有固定作用[3,4]。据估计,在矿质氮含量高的土壤中,大豆种子中共生固结的氮含量为25-50%,而在有机质和氮含量低的土壤中,共生固结的氮含量为80-94%[5,6]。
观察到不同菌株间共生固氮的差异。例如,有几位作者报道,接种日本芽孢杆菌USDA123在无根瘤菌土壤中生长的大豆种子产量低于接种已知固氮效率高的菌株USDA110[7,8,9]。在美国中西部大豆主产区,日本芽孢杆菌USDA123在大田大豆根瘤中的发病率很高[10,11,12,13,14,15],该菌株对根瘤位点的竞争非常激烈[11,16,17]。
为了提高大豆的固氮效率,从早期开始,人们就进行了许多尝试,包括使用USDA110等优良的接种菌株[7,18],改进接种方法[13,19,20],创造固氮能力增强的根瘤菌突变体或转基因菌株[21,22,23],以及选择基因型[16,24,25,26,27]。其中,鉴定用国产血清簇123限制结瘤的大豆材料和开发限制低效固氮菌株的大豆品种是提高大豆共生固氮效率和提高产量的经济有效途径。
美国农业部大豆种质库包括从84个国家引进或在美国开发的18480种栽培大豆和1168种野生大豆[28]。在美国马里兰州贝尔茨维尔的大豆基因组学与改良实验室,用含有52,509个单核苷酸多态性(snp)的SoySNP50K BeadChip试验对这些样品进行基因分型。SoySNP50K数据集极大地促进了对来自不同国家的多种种质资源的筛选,从而发现限制菌株USDA123结瘤的个体或偏好高效固氮菌株结瘤的个体。因此,有可能将本地血清群集123株的结瘤限制和有利于一种或多种有效菌株的结瘤结合到一个基因型中。种质资源和基因型数据集也是通过研究控制结瘤限制和偏好的基因或基因组区域来了解性状复杂性的宝贵资源。其他缓生根瘤菌的结瘤限制基因有Rj2限制日本芽孢杆菌USDA122 [29,30], Rj3限制elkanii芽孢杆菌USDA33, Rj4限制elkanii芽孢杆菌USDA61 [31,32], Rfg1限制Ensifer/Sinorhizobium fredii芽孢杆菌USDA257[33]。此外,携带纯合子隐性等位基因rj1、rj5和rj6的大豆限制了所有根瘤菌菌株[34]。然而,USDA123或USDA110对限制性结瘤的遗传控制尚未见报道。
因此,本研究的目的是确定单次接种日本双歧杆菌菌株USDA110和USDA123后不同栽培大豆和野生大豆组合间根瘤数(NN)的遗传变异,通过对USDA110和USDA123双接种大豆的根瘤占用特性,鉴定可能限制USDA123而倾向于USDA110的新种质,并确定大豆品种中根瘤数性状的遗传复杂性。
分别用日本芽孢杆菌菌株USDA110和USDA123单次接种后,对1321株G. max进行神经网络计数(表S1)。其中两份G. max材料未与两种试验菌株中的任何一种结瘤。它们在田间土壤中生长,证实根部没有结瘤。检测的非结瘤品种为PI548193 (T201),这是一种众所周知的rj1基因型非结瘤品种,以及1987年从日本引入美国的PI507429 (Tousan 89) (www.ars-grin.gov),此前未报道过限制结瘤品种。
接种USDA110时,1321份G. max材料的平均NN值为35.3节/株,范围为0 ~ 75.3节;接种USDA123时,平均NN值为33.8节/株,范围为0 ~ 75节。接种USDA110和USDA123后,大豆单株平均结瘤数分别为12.9和13.8个,分别为5.3 ~ 23.5个和2.3 ~ 27.8个。栽培大豆和野生大豆的神经网络均为正态分布,偏度在- 2 ~ + 2之间,峰度在- 7 ~ + 7之间(表1),这是正态分布的阈值。用USDA110和USDA123接种单株大豆的遗传率分别为0.70和0.68,用USDA110(0.6)和USDA123(0.74)接种大豆的遗传率相似。黄豆和黄豆的神经网络表型变异主要是遗传效应,受环境影响较小,即实验中重复间神经网络一致性较高。
表1不同组的单株结节数(NN)分布g·马克斯和野生大豆单一接种的材料b .日本血吸虫应变USDA110和USDA123
在1319份栽培大豆和69份野生大豆材料中,分别接种USDA110和USDA123,采用t检验(P < 0.05)确定USDA110与USDA123的神经网络差异有统计学意义。未结瘤的品种不包括在本分析中。42份栽培大豆材料USDA110的神经网络显著高于USDA123, 31份栽培大豆材料USDA110的神经网络显著低于USDA123(表S2)。其中,单次接种USDA110高NN和USDA123低NN的栽培大豆品种前10名分别为PI360964、PI594879、PI437367、PI180532、PI603526、PI597471A、PI567564、PI165673、PI548461和PI603308A。相反,USDA110神经网络较低而USDA123神经网络较高的前10个接入是PI240664、PI153231、PI567270A、PI603531A、PI437431、PI464912、PI088448、PI323557、PI054620-2和PI200503(图1)。
此外,5个野生大豆品种PI562558、PI407270、PI522226、PI407318A和PI245331在USDA110结瘤时的神经网络显著高于USDA123,但未发现USDA123结瘤数量高于USDA110的品种。
从单次接种USDA110和USDA123时神经网络差异显著的42个栽培大豆品种中,选择30个USDA110神经网络高于USDA123的品种和5个USDA123神经网络高于USDA110的品种,用USDA110和USDA123混合接种(图1;表S2)。
菌株USDA110在13份材料中对USDA123具有很强的竞争优势。其中,PI567624、PI324187B、PI071465、PI628961、PI189939、PI490766、PI070466-3、PI068731、PI603526、PI438335和PI080671单次接种时USDA110的NN值较高,而引种PI068709和PI464912的USDA123 NN值较高。另一方面,菌株USDA123在5份材料中对USDA110具有很强的竞争能力,其中2份在单次接种试验中对USDA123具有较高的NN (PI153231、PI323557), 3份在同一次接种试验中对USDA110具有较高NN (PI548572、PI378658、PI594879)(图2;表S3)。
在35个加入物中,27个形成了三种不同类型的结节,包括USDA110、USDA123和混合结节(USDA110 + USDA123结节)。其中16个品种的结核占有偏好为USDA110 >混合型结核> USDA123;7个基因型有两种类型的结节:USDA110和混合结节,偏好USDA110, 1个基因型的所有结节都被菌株USDA110占据(图2)。在这些基因型中,一个结节的双菌株占用率从0到67%不等,平均为32.9%。
图1
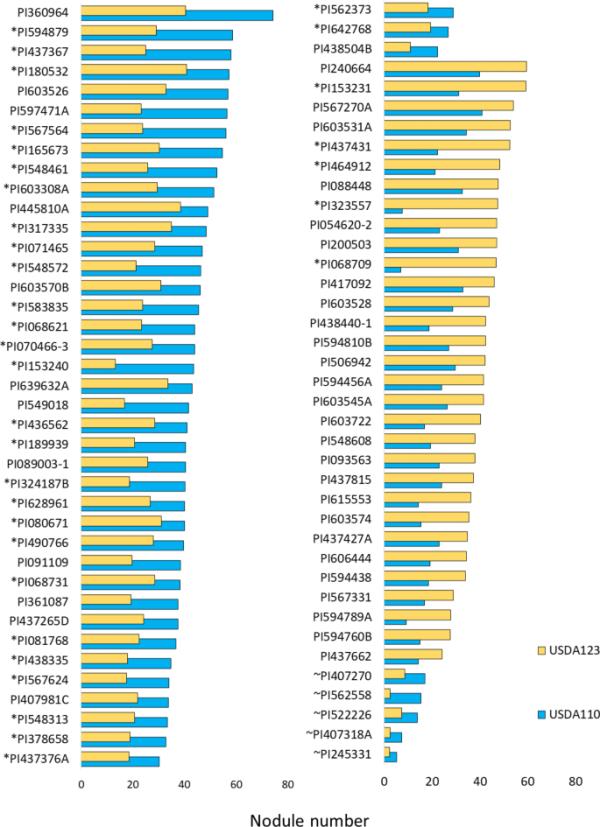
接种日本慢生根瘤菌菌株USDA110和USDA123后,73个G. max和5个G.大豆(加~)材料的根瘤数差异显著(P < 0.05)。选择带星号(*)的品种,分别用USDA110和USDA123混合接种,进行后续竞争试验
图2
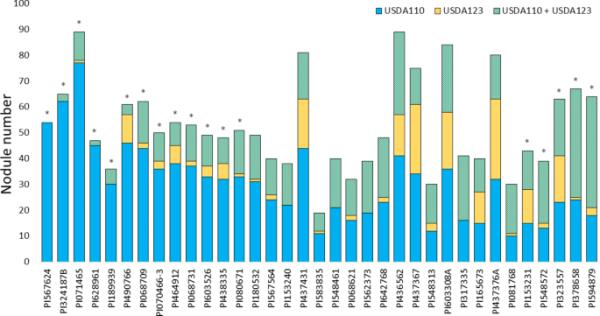
用USDA110和USDA123混合接种的35份大豆材料中含有日本慢生根瘤菌USDA110、USDA123或USDA110和USDA123的根瘤数。星号(*)表示在5%的概率水平上,含有USDA110的结节数与含有USDA123的结节数以及同时含有USDA110和USDA123的结节数有显著差异
基于max和大豆群体k值均为3的最佳拟合模型,大豆群体的cMLM和大豆群体的MLM是控制基因组膨胀的最佳拟合模型(图S1 ~ S4)。此外,全基因组关联研究(GWAS)发现,当单次接种USDA110 (NNmax110)时,共有50个snp与神经网络显著相关(-Log10(p) > 3),当单次接种USDA123 (NNmax123)时,共有50个snp与神经网络显著相关,而单次接种USDA110 (NNsoja110)和USDA123 (NNsoja123)时,分别有32个和51个snp与神经网络显著相关(图3)。
在NNmax110中,50个显著相关的snp中有9个位于染色体14号染色体的连锁不平衡(LD)单倍体块Gm14_Hap123上,全长0.43 Mb(从9到9.5 Mb)。在NNmax123中,50个SNPs中有11个位于11号染色体的正染色质区,长度为1.4 Mb(从0.1到1.5 Mb),位于5个不同的单倍体块中,每个单倍体块中有1到4个SNPs。NNmax110与NNmax123之间仅有4个snp相同,即位于第3染色体11.5 Mb位置的单倍块Gm03_Hap5上的ss715584397 snp;6号染色体13.8 Mb位置的ss715593108;ss715620123在14号染色体上,单倍块Gm14_Hap123;和位于第16染色体33.7 Mb位置的ss715624623。
在NNsoja110中,32个显著snp中有6个位于11号染色体的异色区。6个SNP中有5个位于单块Gs11_Hap28 (25.116-25.123 Mb),另外1个SNP位于下游0.9 Mb。对于NNsoja123,在第17号染色体上发现了21个/ 51个snp,其中6个位于Gs17_Hap5单倍体中,横跨4.2 Mb (22.7-23.7 Mb)的异色区,15个snp位于单倍体中具有1 - 6个显著snp或不具有显著snp的区域。处理NNsoja110和处理NNsoja123之间或种间未发现显著的共同snp。
在NN的最终重要位点计数中,我们保留了重要的先导SNP来代表每个单倍体,以及单倍体之外的SNP。结果,在NNmax110、NNmax123、NNsoja110和NNsoja123中分别检测到35个、35个、23个和34个位点,其中包括NNmax110和NNmax123之间的四个公共区域(表S4)。然而,每个位点只能解释0.8 - 1.5%的表型变异。
图3
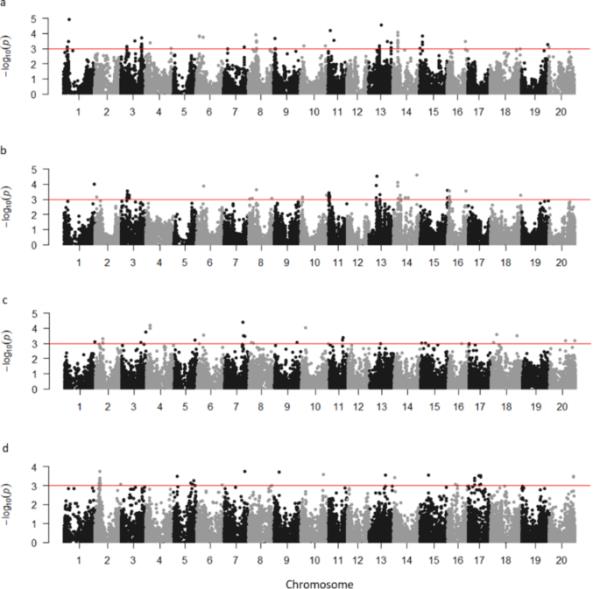
a NNmax110、b NNmax123、c NNsoja110和d NNsoja123在栽培大豆和野生大豆中与根瘤数(NN)相关的曼哈顿区。利用NNmax110和NNmax123的混合线性模型(MLM),以及NNsoja110和NNsoja123的压缩混合线性模型(cMLM),根据20条染色体上的每条染色体的位置绘制了负log10转换的SNPs p值。显著性状相关snp (P < 0.001)高于红色阈值线
摘要
背景
结果
讨论
结论
方法
数据可用性
缩写
参考文献
致谢
作者信息
道德声明
搜索
导航
#####
传统上,科学家使用Leonard Jars作为生长系统来研究豆科植物的结瘤[35,36,37,38,39,40]。后来,一些出版物报道了使用Deepots对大豆囊肿线虫进行实验(Stuewe & Sons, Inc., Tangent, Oregon, USA)[41,42]。Leonard Jars和Deepots是类似的生长系统,其主要优点是生长可控、污染控制、自灌和获得完整的根系进行表型分析。两个系统占用的面积相似,大约为12株/平方米。对于本研究这样的大量样本实验,建立一个有效的、空间最大化的表型系统是保持相似生长条件的基础。因此,164 ml(直径3.8 cm x 21 cm深)的锥形容器(Stuewe & Sons, Inc., tan, Oregon, USA)具有相同的优点,但比Deepots和Leonard Jars小,用于评估大豆根瘤。集装箱以45个单位/托盘为单位分布,面积约为22株/m2,与Deepots和Leonard Jar系统相比,面积增加了83%(图4a)。此外,大豆的入种萌发和生长均匀(图4b)。结瘤取决于几种环境条件,特别是光照、温度和硝酸盐浓度;本研究遵循了之前报道的无硝酸盐培养基中根瘤数表型分型的标准方案[24,43,44,45]。然而,在田间条件下或在温室中与硝酸盐的基质上进行进一步的竞争研究是必要的。虽然之前在三叶草[46]、豌豆[47]和豆科黄芪[48]的结瘤实验中有类似但不完全相同的系统报道,但这是首次在大豆中使用该系统。
同样,在大量材料中确定结核占用率的方法对于本结瘤竞争研究至关重要。本研究建立的快速DNA提取和高通量竞争等位基因特异性(KASP)基因分型方法使我们能够准确区分结核的类型,并通过对结核的Sanger测序和USDA110和USDA123菌株对照验证了这一点。在最初的实验中,使用PrepMan Ultra (Applied Biosystems)进行DNA分离;然而,用KASP方法进行DNA扩增并没有像预期的那样扩增,影响了等位基因的区分,这可能是由于结核的多糖与PCR化学试剂之间的相互作用。众所周知,多糖可以抑制PCR[49,50]。
图4

a Cone-tainers(黄色)和Deepots(黑色)所需面积的比较,b Cone-tainers(黄色)和Deepots(黑色)中栽培大豆根瘤数高通量表型的发展比较
在菌株竞争实验中,首先要确定一种菌株比另一种菌株更倾向结瘤的品种。在本研究中,单次接种USDA110与USDA123的高神经网络首次筛选的30个品种中,有11个品种在竞争试验中对USDA110结瘤的偏好与USDA123相同,而初始选择USDA123与USDA110的高神经网络的5个品种中,有2个品种在竞争试验中具有与单次接种相同的偏好,其余3个品种对USDA110优于USDA123。提示菌株竞争机制的复杂性。在竞争研究中,在控制条件下,混合结节的发病率从0到67%不等,基因型之间差异很大。大豆和其他豆科植物的结节混合感染很常见,以前也有报道[24,25,51,52,53,54,55]。
早期Lindemann等[56]发现,接种两种菌株的大豆在无菌沙中生长的根瘤中,高达32%的根瘤含有混合根瘤。Moawad和Schmidt[57]也描述了类似的结果,双占率为12-32%,Okogun和Sanginga[58]为34%,Payakapong等[44]为4-69%。Lohrke等人[25]报道了一个以上的菌株感染0 - 44%的大豆结节。此外,单个根瘤可能含有不止一种基因型的类菌共生菌,也可能含有非根瘤菌物种[51,53,54]。
为了找到USDA123株结瘤受限的基因型,Cregan和Keyser[26]筛选了适应美国南部成熟群体的材料。他们在栽培品种‘Lee’和选定的受限的22个基因型(1 ~ 8)上计算了每株30个根瘤。在相同条件下,‘李’的单株结瘤数为51个,同时接种3个品种PI371607、PI548461和PI325779时,单株结瘤数分别为35.0、25.8和26.7,说明接种USDA123后,这3个品种的结瘤数小于‘李’。这些作者还发现,与5个标准品种相比,13个基因型的USDA123结瘤受限;将PI371607与栽培品种威廉姆斯(Williams)一起播种在不含日本芽孢杆菌的土壤中,接种USDA123和USDA110、USDA122或USDA138。在每个竞争处理中,超过75%的‘Williams’根瘤被USDA123占据,而在所有情况下,PI371607基因型的根瘤中不到10%含有USDA123。菌株USDA123基因型被排除,而接种菌株USDA123基因型被排除。在本试验中,PI371607接种USDA110(35个瘤/株)和USDA123(37.3个瘤/株)后差异不显著。因此,本研究未将其纳入进一步的竞争实验。然而,当对被Cregan和Keyser[26]分类为USDA123限制结瘤的PI548461 (' Improved Pelican ')进行检测时,发现USDA110(52.5)结节数量明显高于USDA123 (25.8) (P < 0.05)。
通过不同的方法,Cregan等人[24]发现了一种限制USDA110、MN1-1c和USDA138结瘤的基因型。因此,以菌株USDA123和USDA110单次接种PI417566,单株NN分别为31和5。随后,Lohrke等[25]在一个等效的实验中,对同一株的USDA123和USDA110进行了测试,结果相似,每株32个(USDA123)和3.3个(USDA110)结核。本试验中,单次接种USDA123和USDA110时,PI417566单株结瘤数分别为54和32个,但差异无统计学意义。
鉴定具有USDA123根瘤菌潜在结瘤限制和优良根瘤菌特异性的材料,可以通过对材料基因的杂交和渐渗,培育出具有良好共生关系的大豆品种。从本研究中鉴定出几个限制usda123和偏好usda110的材料,可以为进一步的遗传研究和大豆改良提供有价值的资源。
虽然有几篇文章报道了大豆中特定的慢生根瘤菌或Esinfer菌株限制结瘤,但尚未报道发现限制USDA123结瘤的基因[24,25,26,59]。
在本研究中,以日本双歧杆菌菌株USDA110和USDA123为单次接种材料,在NNmax110处理中鉴定出35个位点,在NNmax123处理中鉴定出相同数量的位点。在NNsoja110和NNsoja123中分别鉴定出23个和34个位点。从总共127个位点中,鉴定出107个独特的显著位点,其中18个位点先前在Soybase中报道过NN或结节相关性状(http://soybase.org)。NNsoja123中位于第5染色体31.4 Mb的显著SNP ss715590625与Glyma.05G121600 (GmVTL1a/ nodin -21)基因相邻,该基因是根瘤共生固氮不可或缺的液泡铁转运蛋白样基因,可将亚铁从受感染的根细胞胞浆转运到共生体[60,61,62]。
Yang等人[63]在6号染色体上发现了两个位点,分别是NNmax110中的ss715595435 (6581,862 bp)和NNmax110和NNmax123中的共同SNP ss715593108 (13759,357 bp),位于6529,355至16,221,141 bp的区间位置(根据Wm82.a.2的位置)。v1, Song等人[64],这与Hwang等人[65]对NN描述的区域(12,336,655至14,225,129 bp)相似。另一方面,Nicolás等[66]在同一6号染色体上发现了一个位于44,884,659 ~ 49,271,843 bp区域的QTL,其中SNP ss715594793 (47,869,389 bp)对NNsoja123具有显著性。
在10号染色体上,在NNmax110中的单倍块Gm10_Hap93中的ss715607620 snp (43,688,879 ~ 46,747,178 bp)和NNmax123中的ss715607699 snp (47,626,066 bp)在Yang等[63]先前报道的6个QTLs根瘤相关性状的相似区域(43,509,883 ~ 48,424,003 bp)被发现。
在11号染色体上,Yang等人[63]和Santos等人[67]分别发现了NN的显著区域(区间分别为2,718,971至6,217,092 bp和4,234,240至9,107,006 bp),其中SNP ss715610750 (4,611,787 bp)在NNmax110中显著。此外,Nicolás等人[66]在14号染色体的广泛区域发现了两个与NN和单株结节干重有关的qtl,从16,249,052到45,041,580 bp,其中在NNmax123中发现了三个显著snp, ss715617892位于22,010,302 bp, ss715618045位于单倍块Gm14_hap29(26,079,931至26,911,748 bp), ss715618900位于43,535,411 bp。在第15号染色体上,Hwang等人[68]和Santos等人[67]分别描述了4,434,024至5,540,983 bp和7,537,816至43,037,732 bp的结节大小区域,其中发现了三个显著SNP,分别是NNmax110的单倍块Gm15_Hap51(5,218,552至5,252,046 bp)的ss715622828 (5,241,013 bp), NNsoja110的ss715620209 (9,445,842 bp)和NNsoja123的SNP ss715621072 (16,434,045 bp)。
在NNmax110和NNmax123中,16号染色体上的SNP ss715624623是4个显著相关的共同标记之一,其中有7个推测基因存在。其中Glyma.16G175600 (GmUGT1)、Glyma.16G175900 (GmUGT7)和Glyma.16G176000 (GmIF7GT5)三个位点与共生有关,参与编码异黄酮7- o -葡萄糖基转移酶的异黄酮途径,该途径在植物与微生物相互作用中起重要作用,是与日本芽孢杆菌建立共生关系所必需的[69]。此外,异黄酮染料木黄酮和大豆苷元也是日本蓟(B. japonicum) nod基因表达的主要诱导剂[70,71,72],但GO分析后未发现显著项。
在19号染色体上,位于49,239,109 bp的SNP ss715635907在NNmax110中具有显著性,并且在结节干重的49,210,133至49,243,536区域也被Nicolás et al.(2006)描述。在20号染色体上,在NNsoja123中发现的两个显著SNP,分别是Gs20_Hap55单倍块中的ss715638590 (44,971,784 bp至44,992,638 bp)和ss715638603 (45,139,632 bp),此前已被Hwang等人描述[68];在NNsoja110中发现的一个显著SNP, ss715637162 (29,015,399 bp),位于扩展区域(2,717,137至34,051,592 bp),由Santos等人[67]描述根瘤干重。此外,在4个处理中,NNmax110和NNmax123处理有4个显著位点重叠,即3号染色体11.6 Mb位置的snp ss715584397,单倍体块Gm03_Hap5;6号染色体13.8 Mb位置的ss715593108;ss715620123在14号染色体上,单倍块Gm14_Hap123;和位于第16染色体33.7 Mb位置的ss715624623。
检测到多个与NN相关的qtl,接种处理之间共有或重叠的qtl数量较少,每个位点的影响较小,这表明该性状的复杂性以及根瘤菌与宿主之间建立共生关系的复杂信号交换。该研究也支持了寄主控制的限制或结瘤偏好是菌株依赖的证据。
位于美国伊利诺伊州厄巴纳-香槟市的美国农业部大豆种质库(https://www.ars-grin.gov/)提供了18,480份max和1168份大豆种质,之前使用SoySNP50K测定法进行了基因分型,其中包含超过50,000个snp[28]。对数据集的分析表明,4303个G. max(23%)和362个G. soja(30%)与该集合中的另一个成员至少99.9%相同。从剩余的14186份栽培大豆和806份野生大豆材料中,选择1417份栽培大豆和81份野生大豆材料,采用以下方法:通过计算不同等位基因的snp数与配对材料间总位点数的比值,估算14186份栽培大豆和806份野生大豆材料间的配对距离。栽培大豆和野生大豆分别聚在1418个和82个聚类中,约占收集的栽培大豆和野生大豆材料的10%。从每个聚类中选择一个与其他聚类平均距离最大的聚类。进一步分析表明,根据所选材料中多态性snp数与所有栽培大豆和野生大豆材料中多态性snp总数的比值,这两组可捕获栽培大豆和野生大豆材料中90%以上的遗传多样性。然而,由于某些品种的种子缺乏或不足,本研究仅使用了1418个大大豆品种(地方品种和现代品种)中的1321个品种和81个大大豆品种中的69个品种(表S1)。这些加入代表了51个国家和所有成熟度组(000至X)。
这些植物是在美国马里兰州美国农业部贝尔茨维尔农业研究中心的温室中种植的。所有实验条件相同,自然光补充白炽灯,光周期为16 h,温度范围为25±7°C。164毫升(直径3.8厘米x 21厘米深)的容器(Stuewe & Sons, Inc., Tangent, Oregon, USA)用于种植植物。在每个集装箱的底部放置一个中等大小的棉球,里面装满了预洗过的沙子。容器由162个细胞(9 × 18)的幼苗单元支撑,每组45个单元分布,每个容器之间留下一个空细胞(5列x 9行)。苗木和容器被放置在装满3英寸自来水的托盘中。沙子和棉球被高压灭菌,容器、平板和托盘在1:4的漂白剂溶液中消毒5分钟,然后用水冲洗三次。为了消毒和播种,将162个细胞的幼苗平面改造成分区过滤器。首先,将尺寸缩小到45个单元格(5 × 9单元格),并在每个单元格的底部粘上塑料网# 10。然后,将45个品种的种子和塑料标签分布在种子过滤器上,依次浸泡种子过滤器消毒,先用70%乙醇在托盘中浸泡30秒,然后用水冲洗,在另一个托盘中用1:4的漂白剂洗涤4分钟,最后用水冲洗4次,逐个托盘转移种子过滤器。种子过滤器的分布与实验装置的孔-容器相同。因此,每次消毒一次使用四个种子过滤器。用该方法对180个品种的种子进行了6 min的消毒。
试验采用完全随机区组设计,选用黄豆品种1321个,黄豆品种69个,分4个重复。每个重复,每个容器播撒4粒种子,4天后,将幼苗稀疏至1粒,并用无氮Hoagland-Arnon植物营养液湿润[73]。分别用2 ml (108 CFU/ml-1)的日本芽孢杆菌USDA110和USDA123悬浮液单次接种5日龄植株。托盘里的水每周换一次。30天后,收获根,清洗,干燥,冷冻,然后除去根瘤并计数。36株未接种的“Williams 82”植株作为对照,沿长凳分成4个单元,表型分析时未发现根瘤。
采用4个重复的完全随机设计试验,35份材料分别接种USDA110和USDA123后,其神经网络差异显著。用USDA110和USDA123等浓度(108 CFU/ml?1)、体积(1:1)的2 ml混合悬浮液对5日龄植株进行二次接种。30天后,收获根,清洗,用75%的酒精消毒,在28°C下干燥20分钟,然后冷冻以作进一步分析。之后,将根瘤从根部分离,置于无菌96孔板(0.8 ml)中,每孔1个根瘤,冻干过夜,提取DNA。在每个孔中放置一个2.4 mm的钢珠,并用热铝膜密封板。结核在Retsch MM400混合器中以30 Hz的频率研磨30 s。皿在5200 rpm下离心5 min,每个样品加入500μl蒸馏水。将上清液100μl转移到新的96孔PCR板上,密封,5200 rpm离心10 min;在不干扰球团的情况下取下上清,然后在球团中加入50μl蒸馏水,短暂旋转板,然后在100℃下加热10 min,在3000 rpm下离心5 min,最后取上清20μl,转移到新的96孔板中。将DNA稀释至10%进行PCR反应。
为了确定双接USDA110和USDA123后大豆中USDA110和USDA123的根瘤占用率,基于USDA110 (https://www.ncbi.nlm.nih.gov)和USDA123 (http://genome.jgipsf.org)的基因组序列,利用基于WASP的基于web的等位基因特异性引物设计工具(https://bioinfo.biotec.or.th/WASP)[74]设计了一组23个竞争等位基因特异性PCR (KASP)标记。其中,rhi_snp2(等位基因-1正向引物序列:gaaggtgaccaagttcatgctccaggatgatcacgccc,等位基因-2正向引物序列:gaaggtccaacggattccaggatgatcacgccg,反向引物序列:gcacaggtcctcctactg)和rhi_snp24(等位基因-1正向引物序列:gaaggtgaccaagttcatgctgtggtcgttgctggct,等位基因-2正向引物序列:gaaggtcggagtcaacggattgtggctcgttgctggcc和反向引物序列GACAACATGATCCTCGCGCTC)的扩增效果和等位基因识别效果最好。因此,选择这两种标记对35份G. max材料进行根瘤占用率测试。用KASP标记对结核进行基因分型,最终反应量为5μL,与2.5μL的2x KASP MasterMix Low Rox 5000 V4.0 TF (LGC Genomics)、0.07μL的引物混合物(每种正向引物12μM加反向引物30μM)和5 - 20 ng基因组DNA混合。使用QuantStudio 6 Flex (Applied Biosystems, Thermo Fisher Scientific)在以下循环条件下进行PCR,在94°C下活化15分钟;在94°C下20 s和61-55°C下60 s的10个循环(每个循环的退火温度每循环降低0.6℃);在94℃和55℃分别进行26次20 s循环和60 s循环,然后在94℃和57℃分别进行15次20 s循环和60 s循环。在35°C下运行30 s后进行荧光检测,并使用Quant Studio Real-Time PCR软件(Applied Biosystems, Thermo Fisher Scientific)分析数据。为了验证表型结果,从3个混合结核中提取了12、13和18个类杆菌,在慢生根瘤菌多位点序列分析中定义的7个位点进行了sanger测序[75]。采用t检验确定了USDA110和USDA123在大豆种植过程中根瘤占用率的差异。
用日本双球菌菌株USDA110 (NNmax110)和USDA123 (NNmax123)单株和USDA110 (NNsoja110)和USDA123 (NNsoja123)单株接种的大豆进行遗传变异和遗传力分析。为了确定神经网络的复杂性,通过全基因组关联研究(GWAS)分析了位点的数量及其对性状的影响。采用ADMIXTURE v1.22[76]软件分析种群结构,将G. max和G. soja的K值分别设置为2 ~ 10和2 ~ 5,并对每个K值分别设置30、100、500、1000和2000粒种子进行10倍交叉验证(CV)。最可能的k值是用CV值确定的。因此,在Tassel[77]中,使用考虑亲缘矩阵和种群结构的一般线性模型(GLM)、规则混合线性模型(MLM)和压缩混合线性模型(cMLM)测试了标记和神经网络的关联。snp -性状关联的显著性阈值设为经验值P 3。在Haploview 4.2中确定了大豆基因组的单倍型块[78],并将显著qtl与Soybase (http://soybase.org)和先前报道中发现的NN和其他根瘤相关性状的qtl进行了比较。当给定的QTL位于先前描述的区间位置之外时,我们将其视为新QTL。在Phytozome (http://k1.fpubli.cc/file/upload/202309/09/nshudprvvq0)中,寻找位于显著区域的基因在结节相关性状中的表达丰度。[79],保留根瘤菌介导结瘤的相关基因。最后,在agriGO toolkit v.2.0 (http://systemsbiology.cau.edu.cn/agriGOv2)中对这些基因进行基因本体(GO)分析[80]。在候选基因少于10个的情况下,我们添加了一些随机基因,使程序能够进行GO分析。
以下是电子补充材料的链接。
下载原文档:https://link.springer.com/content/pdf/10.1186/s12864-023-09627-4.pdf

